Täheldatud ühel igal 100 000 vastsündinul, on Pfeifferi sündroom seotud FGFR1 ja FGFR2 geenide mutatsiooniga; mõlema geeni ülesanne on reguleerida koljuõmbluste sulandumist ning sõrmede ja varvaste arengut.
Pfeifferi sündroomi diagnoosimiseks on füüsiline läbivaatus, anamnees, kolju ja sõrmede ja varvaste radioloogiline hindamine ning lõpuks geneetiline test.
Praegu saavad Pfeifferi sündroomi all kannatavad inimesed loota ainult sümptomaatilisele ravile, st sümptomite leevendamisele.
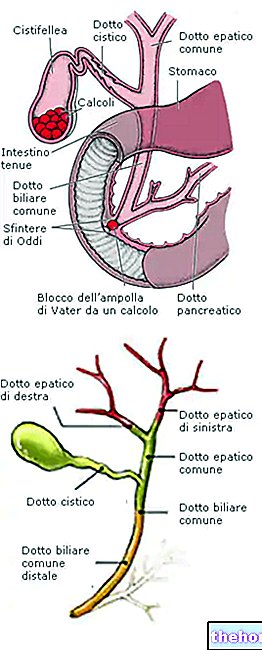
Lühike ülevaade koljuõmblustest ja nende sulandumisest
Kraniaalõmblused on kiulised liigesed, mis ühendavad kraniaalvõlvi luid (st eesmise, ajalise, parietaalse ja kuklaluu).
Normaalsetes tingimustes toimub koljuõmbluste sulandumisprotsess sünnijärgsel perioodil, alustades mõnede liigeste elementide puhul 1–2-aastasest ja lõpetades 20-aastaselt. See pikk ja järkjärguline sulandumisprotsess võimaldab ajul piisavalt kasvada ja areneda.
- Ebanormaalselt suurte ja kõrvalekaldunud pöidlate ja suurte varvaste olemasolu selliselt, et need näivad teistest varvastest eemalduvat (mediaalne kõrvalekalle).
Seetõttu on Pfeifferi sündroom geneetiline seisund, mis määrab selle kandjatel peamiselt kolju ja käte anomaaliad.
Kuna lugejatel on võimalus sümptomitest pühendatud peatükis rohkem teada saada, võib Pfeifferi sündroomi seostada muude probleemide ja muude füüsiliste väärarengutega.
Epidemioloogia: kui levinud on Pfeifferi sündroom?
Statistika kohaselt sünnib iga 100 000 inimese kohta Pfeifferi sündroom.
Kas teadsite, et ...
Geneetilised haigused, mis nagu Pfeifferi sündroom põhjustavad kraniosünostoosi, on umbes 150.
Nende hulgas paistavad lisaks Pfeifferi sündroomile silma Crouzoni sündroom, Aperti sündroom ja Saethre-Chotzeni sündroom.
Mis põhjustab Pfeifferi sündroomiga seotud geenimutatsiooni?
Eeldus: inimese kromosoomidel esinevad geenid on DNA järjestused, mille ülesanne on toota eluks olulistes bioloogilistes protsessides, sealhulgas rakkude kasvus ja replikatsioonis, põhilisi valke.
Kui nad on mutatsioonivabad (seega tervetel inimestel), toodavad FGFR1 ja FGFR2 geenid õiges koguses vastavalt Fibroblasti kasvufaktori retseptorit 1 ja fibroblasti kasvufaktori retseptorit 2, mis on kaks retseptorvalku, mis on olulised kraniaalsete õmblusniitide ajastus ning sõrmede ja varvaste arengu reguleerimine (teisisõnu, need annavad märku, millal on sobiv aeg koljuõmbluste liitmiseks, ning kontrollivad sõrmede ja jalgade teket).
Teisest küljest, kui nad läbivad Pfeifferi sündroomi juuresolekul täheldatud mutatsioone, on FGFR1 ja FGFR2 geenid hüperaktiivsed ja toodavad ülalnimetatud retseptorvalke nii suurtes kogustes, et koljuõmbluste sulandumisajad muutuvad (need on kiiremad) ) ning sõrmede ja varvaste treenimise protsess ei toimu õigesti.
Pfeifferi sündroom on autosoomne domineeriv haigus
Aru saama...
Iga inimese geeni esineb kahes eksemplaris, mida nimetatakse alleelideks, millest üks on ema ja teine isapoolne.
Pfeifferi sündroomil on kõik autosomaalse domineeriva haiguse tunnused.
Geneetiline haigus on autosomaalselt domineeriv, kui seda põhjustava geeni ühe eksemplari mutatsioon on piisav avaldumiseks.
Pfeifferi sündroomi tüübid
1993. aastal avaldas Ameerika arst Michael Cohen pärast arvukaid uuringuid Pfeifferi sündroomi kohta kõnealuse geneetilise haiguse tüpoloogilise klassifikatsiooni, mis ennustas kolme patoloogilise variandi olemasolu, mis identifitseeriti lihtsalt terminitega "I tüüp", "II tüüp" ja Tüüp III "ja kõigil on kraniosünostoos ning pöidla ja suurte varvaste anomaaliad. Meditsiiniteaduslik kogukond kiitis selle klassifikatsiooni kohe heaks ja sellest ajast alates on Pfeifferi sündroomi eksperdid kasutanud seda diagnostilise vahendina ja olemasoleva geneetilise seisundi tõsiduse hindamiseks; tegelikult tuleb märkida, et dr Coheni klassifikatsioon eristab Pfeifferi sündroomi kraniaalsete ja digitaalsete anomaaliate raskusastme ning muude sümptomite ja tunnuste olemasolu põhjal.
Üksikute patoloogiliste variantide üksikasjadesse süvenedes on artikli selles punktis oluline rõhutada järgmist:
- The I tüüp. see on Pfeifferi sündroomi vähem raske versioon, kuna kraniostenoos ning pöidla ja suure varba kõrvalekalded on piiratud tagajärgedega.
Muu oluline teave: see on tingitud FGFR2 mutatsioonist, mõnikord kombineerituna FGFR1 mutatsiooniga; see võib olla pärilik või omandatud seisund. - The II tüüp see on Pfeifferi sündroomi kõige raskem versioon, kuna seda seostatakse raske kraniosünostoosiga, mis on eluga peaaegu kokkusobimatu, ning käte ja jalgade sügavate kõrvalekalletega.
Muu oluline teave: see on tingitud ainult FGFR2 mutatsioonist; see on alati omandatud seisund. - The III tüüp just Pfeifferi sündroomi versioon langeb raskusastmel veidi alla II tüübi, kuid tublisti üle I tüübi, kuna praegune kraniosünostoos on peaaegu sama raske kui eelmises punktis kirjeldatud variandil.
Muu oluline teave: see on tingitud ainult FGFR2 mutatsioonist; see on alati omandatud seisund.
Kranioostenoos
Pfeifferi sündroomi kandjatel võib kraniosünostoos sõltuvalt varases sulandumisprotsessis osalevate kraniaalsete õmbluste arvust põhjustada järgmisi tagajärgi:
- Pea täiesti ebanormaalne vertikaalne areng koos kolju külgmise laienemise puudumisega. Seega on Pfeifferi sündroomiga patsiendil pikk ja kitsas pea;
- Kõrge ja silmapaistva lauba moodustamine;

- Suurenenud intrakraniaalne rõhk, millest sõltuvad sellised sümptomid nagu püsiv peavalu, nägemishäired, oksendamine, ärrituvus, kuulmisprobleemid, hingamisprobleemid, vaimse seisundi muutused, papillödeem;
- Intellektuaalsed puudujäägid, mis vähendavad IQ -d. Intellektuaalsed puudused tulenevad aju vähenenud kasvuruumist pärast koronaalsete kraniaalõmbluste enneaegset sulandumist;
- Näo vaheosa vähene areng, mis tundub lame, kui mitte nõgus;
- Punnis (proptoos), laiad silmad ja ebanormaalselt asetsevad silmad (silma hüpertelorism);
- Nokaga nina olemasolu;
- Lõualuu väljaarenemise ebaõnnestumine (ülalõualuu hüpoplaasia), mille tulemuseks on ülerahvastatud hammaste seisund;
- Pea ristikulaadne välimus ("ristiku kolju"). "Ristiku kolju" põhjustab vesipea.
I TÜÜP
I tüüpi Pfeifferi sündroomi seostatakse kerge kliinilise kraniosünostoosiga, mis piirdub sageli kolju pikliku kuju andmisega ning nähtavalt kõrge lauba ja lameda näo tekitamisega.
Õige ravi korral elavad I tüüpi Pfeifferi sündroomiga inimesed tavaliselt normaalset elu ja neil on normaalne IQ.
II TÜÜP
II tüüpi Pfeifferi sündroom on ainus patoloogiline variant, mis põhjustab nn "trefoil kolju", see kraniaalne anomaalia mõjutab tõsiselt intellektuaalseid võimeid ja on sageli seotud enneaegse surmaga.
II tüüpi Pfeifferi sündroomi all kannatavad patsiendid esitavad kogu ülalkirjeldatud kliinilise pildi kraniosünostoosi tagajärgede kohta.

III TÜÜP
III tüüpi Pfeifferi sündroom avaldab kandjatele sama mõju kui II tüüpi Pfeifferi sündroom, välja arvatud "trefoil kolju".
III tüüpi Pfeifferi sündroomiga inimestel pole pikka eluiga.
Pöidlad ja suured varbad mõjutavad kõrvalekalded
Kui pöidlad ja suured varbad mõjutavad kõrvalekalded on eriti tõsised, võivad need tõsiselt kahjustada käte ja jalgade funktsionaalset võimekust, põhjustades esemete haaramise ja / või kõndimise probleeme.
Kas teadsite, et ...
Pfeifferi sündroomiga patsientide pöidlaid ja suuri varbaid mõjutav keskmine kõrvalekalle on näide varus varus'est. Täpsemalt räägivad arstid pöidla varusest, mis on tingitud pöidlate mediaalsest kõrvalekaldumisest, ja hallux varus'est, mis on tingitud suurte varvaste keskmisest kõrvalekaldumisest.

Brahüdaktiliselt
Pfeifferi sündroomi korral on brachydactyly üsna tavaline anomaalia, mis võib mõjutada vaid mõnda sõrme või kogu käte ja / või jalgade digitaalset kompleksi.
Brahüdaktilisuse probleem on täheldatav kõikides tüpoloogilistes variantides, kuigi erineva sagedusega.
Sündaktiilselt
Pfeifferi sündroomi korral kujutab syndaktilia endast üsna sagedast anomaaliat (harvem kui brahüdaktiline), millel võib olla erinev varjund (see võib olla mittetäielik, täielik, keeruline jne).
Brahüdaktilisuse probleem on täheldatav kõigis Pfeifferi sündroomi tüpoloogilistes versioonides, ehkki erinevate kordustega.
Luu anküloos
Pfeifferi sündroomi seostatakse ennekõike küünarliigese luu anküloosiga, kuigi tegelikult võib see põhjustada sama probleemi mis tahes inimkeha suurele liigesele.
Luu anküloos on probleem, mida leidub ainult Pfeifferi sündroomi kõige raskemates tüpoloogilistes versioonides (eriti II tüübi puhul).
Hingamisteid mõjutavad kõrvalekalded
Pfeifferi sündroomist põhjustatud võimalikud kõrvalekalded hingamisteedes võivad põhjustada hingamisprobleeme, millel on tõsised tagajärjed patsiendi üldisele tervisele (aju kannatab kõige rohkem).
Nagu luu anküloos, on ülaltoodud kõrvalekalded täheldatavad ainult raskemates tüpoloogilistes variantides (eriti II tüüpi).
Millal on võimalik Pfeifferi sündroomi tuvastada?
Tavaliselt ilmnevad Pfeifferi sündroomist tingitud kolju- ja digitaalsed kõrvalekalded sündides, seega on diagnoosimine ja ravi planeerimine kohene.
pea (pea röntgen, pea CT ja / või pea MRI) ning käed ja jalad; lõpuks lõpeb see geneetilise testiga.
Füüsiline läbivaatus ja haiguslugu
Füüsiline läbivaatus ja anamnees seisnevad peamiselt patsiendi sümptomite täpses hindamises.
Pfeifferi sündroomi kontekstis tuvastab arst diagnoosimisprotsessi nendes faasides kraniostenoosi ja pöidlaid ja suuri varbaid puudutavad kõrvalekalded ning püstitab teiste olemasolevate sümptomite põhjal käimasoleva tüpoloogilise variandi.
Pea, sõrmede ja varvaste radioloogilised uuringud
Pfeifferi sündroomi kontekstis
- Arst kasutab pea radioloogilisi uuringuid, et kinnitada koljuõmbluste varajase sulandumise olemasolu ja hinnata kolju-aju anomaaliate tõsidust.
- Radioloogilised uuringud on seevastu hädavajalikud, et uurida varususe ulatust ja "võimalikku brahüdaktilisust ja / või" võimalikku sündaktüüli.
Geneetiline test
See on DNA analüüs, mille eesmärk on tuvastada kriitiliste geenide mutatsioone.
Pfeifferi sündroomi kontekstis kujutab see kinnitavat diagnostilist testi, kuna võimaldab esile tõsta FGFR2 ja / või FGFR1 mutatsiooni.
Geneetiline test on ka test, mis võimaldab kindlaks teha olemasoleva Pfeifferi sündroomi tüübi.